【转】中国是否已从仿制药大国变身为创新药大国?
发布时间:2018-04-10
(文章转自公众号“药渡”)
2017年4月,华尔街日报发表题为《China Emerges as Powerhouse for Biotech Drugs》[1],认为中国正在成为国际生物技术药物发展的中坚力量。近几年来,我国医药研发增速已经跃升全球第一,创新产出和能力快速提高。新药研发领域的投资热度也空前高涨,国内也不断涌现出一批创新研发实力派企业。但是总体来说,中国要成为国际生物技术药物开发的主角,差距还是不小。这从每年CFDA批准进入临床和上市的新药种类和数量就能证明现有的药品研究和生产的现状与欧美相比存在不小的差距。中国现有的主流药品仍以化药和中药为主,在全球发展最为迅猛的生物药物领域,所占市场份额却很小。监管部门和医药企业一直在寻找能够快速赶上世界潮流的发展之路。受经济实力有限、基础研究薄弱、企业数量多、规模小、相关科研人才储备不足及相关体制机制不完善等诸多因素的制约,中国医药产业要赶上世界前沿绝非易事。但是,近十年,中国生物药物产业的迅猛发展,为将来中国医药领域赶上世界提供了一个很好的契机。
1、生物药物在现有中国药品市场的地位与世界主流脱节
2015年度中国药品销售总额达到1.33万亿[2]。但是具体去分析比较国内外的药品销售数据,就会发现中国药品的销售状况是与世界完全不一样的。表1比较了2014年度全球/中国销售前10位的处方药品数据,可以看出两者巨大差异。在中国排名前十的畅销药榜单中看不到一个生物药物。反观世界主流市场,其榜单中生物药物已经占据了主要地位。进一步去比较具体产品,可以看出两个市场的一些重要区别:
1.1 国内外畅销药物品种差异巨大
两个榜单中没有一个相同的产品。中国畅销榜中,没有一个现代概念的生物药物。输液溶媒氯化钠,葡萄糖输液分别排在了第1,8位。申捷(神经节苷脂),欧兰同(奥拉西坦)这些产品在国外几乎查不到销售数据。而全球畅销药榜单中,有7个为重组表达的生物药物(6个为单抗,一个为重组蛋白)。
1.2 国内外畅销药物适应症存在差异
国内畅销药榜单30%产品属于明确临床适应症的产品(氯化钠,葡萄糖输液和白蛋白);消化系统用药占据了三个(泮托拉唑钠,奥美拉唑,兰索拉唑),神经内科用药占据了两个(申捷,欧兰同);没有一个抗肿瘤药物。而国外畅销品种集中于肿瘤(3个)、RA(3个)。只有降血脂这个适应症两个榜单中有类似产品(立普妥和Crestor)。国内外畅销品种适应症的差异,肯定不是人种或发病率差异所致,同属亚洲的日本药品销售排名就与世界市场非常相似的。
1.3 国内外畅销药物销售价格迥异
国内这些畅销品种单价均不高,一般单次用药均仅在几十到几百RMB之间,一般是以量取胜;国外品种却是价格昂贵,象Humira、Herceptin这些单抗药物单价都是数千美元,而索非布韦(Sovaldi)上市当初更是以84000美元/疗程的价格吓到了见多识广的美国人。
1.4 技术含量和临床价值差异
全球榜单里十个产品全部为独家专利保护产品,基本在世界主要市场均已上市。这些药疗效显著,甚至达到了“药到病除”的效果,因此虽然价格高昂,但是患者和保险机构还得咬牙承受。国内这些畅销药物,要么专利早就过期,要么根本没有真正意义的知识产权保护。还有一个值得回味的现象:国外的畅销药品,每到一个销售年度结束,都会赶紧披露上年度的销售数据来展现自己的“战绩”;但是在中国获得很多畅销药物具体的销售数据是一件极其困难的事情。向来高调的国人在药品销售额这个领域极其谦虚,很少能见到这些畅销药厂家主动披露年度销售额。中国厂家这么低调的原因又是什么呢?
因此,仅仅从比较生物药物在国内外销售现状就可以发现中国的药品市场与世界主流相比是严重脱节的,甚至是“畸形”的。而从患者角度来说是极其不公平的。患者迫切需要的安全、有效的很多生物药物并没有在临床广泛使用;而一些安全无效的品种,临床收益有限,不解决患者根本问题,但通过诸多商业手段达到了巨额销售。社会和患者付出了大量金钱,但是并没有得到相应的临床收益回报。
2、生物药物将是未来中国医药行业的重要发展方向
2.1 畸形的药品市场不会是中国未来的发展方向
中国药品市场这种“畸形”的现象不可能一直存在。近几年从政府法规到CFDA的各种改革,已经显示“矫畸”行动正在进行中。为患者提供安全、有效、可及的药物一直是政府和社会的迫切期望和要求。中国的药品市场必然也将与世界主流一样:安全有效的药品必将成为市场主流,那些仅仅依靠营销手段达到畅销的产品必然会被市场所淘汰。在强调药物临床价值的大前提下,国内的药品市场,必将从传统的化药、中药为主,逐渐向化药、生物药为主的方向转换。而生物药在国内起点低,基数小,更将是未来的重要方向。
2.2 生物药物已是全球药品市场的重要组成部分
在国际市场,生物药物早已经成为全球药品市场最重要一部分。据First Word Pharma发布的2015年全球品牌药品销售额排名数据,排名前十的销售总额超过800亿美元,生物药物占据了其中八席。排名榜首的Humira达到了140亿,而到了2016年更是达到了惊人的160亿美元。但在中国,即使到2016年,尚未有一个生物药物销售排名进入前十。但是随着国内医药市场的规范,临床价值逐渐成为评价药物的核心,这种与世界药品市场相脱节的现象不可能永远持续,高价有效的生物药物必将逐渐淘汰那些安全无效的制品。
2.3 生物医药领域的人才优势有利于国内企业赶上世界前沿。
生物药物的开发,最重要的一个因素就是人才,而中国刚好在这领域具有一个全球独特的优势。自上世纪八十年代开始,诸多中国学子依靠国外大学提供的奖学金,纷纷出国留学,而最容易获得奖学金的领域就是生命科学,因此相当数量的留学人员选择了生命科学作为留学的起点,而生命科学领域博士毕业后最有“前景”和“钱景”的选择就是去国外大药企做研发。经过30多年的积累,已经在生命科学和生物药物研发领域积累了一批相当数量的华人科学家。随着国内经济实力的发展以及对创新药开发,越来越多的相关人才纷纷回流。“风险投资+海归”或“大药企+海归”这两种模式已经成为国内生物药物开发的典型模式。无论是已被列为创新药标杆的“康柏西普”、“埃克替尼”的开发,还是苏州信达、药明生物、百济神州这些明星生物医药研发企业,都是由海归作为中坚力量。这些海归科学家均有长期在国外从事生物医药的研究经历,他们的大量回归,使得国内生物医药研发的关键技术迅速赶上了世界前沿。例如抗体高通量筛选、CHO高表达、单抗药物的大规模生产等技术,在十年以前在国内还一直被视为极具挑战的技术,现在已经成为所有生物药物研发企业的标配。在中国获得一个生物仿制药申报临床的全套技术,已经跟去饭店点菜那么简单。仅仅一个阿达木单抗,就已经有23个企业进入临床或递交了IND申请。这里面的进步大部分要归功于这些海归人员。
2.4 生物药物的结构特点更有利于开发follow-on产品
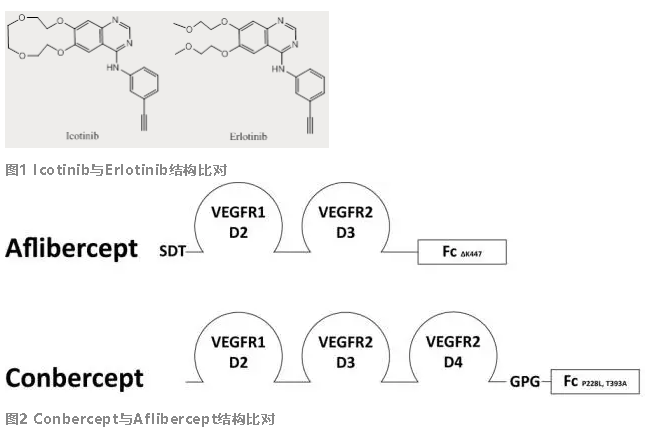
小分子药物结构比较简单,在现有分子基础上进行改造修饰,最终对活性、选择性、生物利用度、DMPK、药效/安全性等的影响很难预测。要筛选获得一个既能避开专利保护、临床疗效又不劣于被仿产品的新化合物绝非易事。但是生物药物,特别是抗体类药物,虽然结构复杂,但是只要靶点或表位没有被保护,在原来序列基础上做一些简单的改进,甚至重新筛选一个me-too类抗体,对大部分中国公司来说已并非难事。埃克替尼和康柏西普这两个创新药完美地体现了这个差异。埃克替尼(Icotinib)为了绕开专利保护,在Erlotinib基础上进行了结构改造(图1),差别仅在侧链的开环与闭环。虽然绕开了专利限制,但实际上其PK和活性都有所下降,从Erlotinib的每天一次(150mg/次)变成了埃克替尼的每天3次(125mg/次),从临床角度看是一个变劣改造。而同期成都康弘开发的生物创新药康柏西普,也是为了绕开专利在Aflibercept基础上增加了VEGFR2 第4个结构域的部分序列[3](图2),但是两者临床疗效没有显著差异。甚至康柏西普的给药方案在Aflibercept基础上进行了优化,达到了更少的给药频率而维持相似的临床疗效[4]。因此虽然同是me-too类药物,但是临床疗效上的不同导致上市后的走向逐渐产生较大的差异。早在2011年上市的埃克替尼一直只能靠政策和价格优势在国内攻城略地,很难走出国门;而2014年才上市的康柏西普不仅逐渐占据了国内市场,同时也在2016年10月在美国开始了III期临床,准备走向国际市场。
3、具有临床价值的创新生物药的开发将是未来中国生物药物领域的主旋律
早在二十多年前,药企、资本、政府都已经相信生物药物是中国医药产业未来发展的一个重要方向。从早期的细胞因子热、单抗热、ADC热到现在的肿瘤免疫治疗热,一个接一个热潮一直在国内交替涌现,但是未来只有那些真正具有临床价值的创新生物药才会是中国医药市场的主流。
3.1 生物仿制药的开发热潮已过
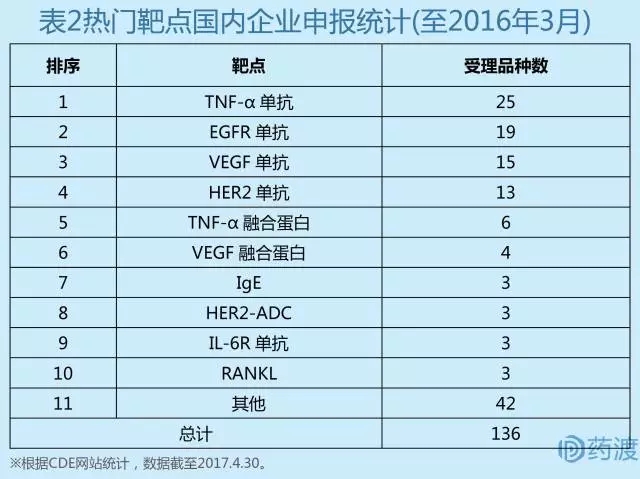
由于国外巨额销售的示范效应,十多年前在中国涌起了一股生物仿制药,特别是仿制单抗的热潮。另一方面,随着国内技术能力的提高,单抗药物的生产工艺壁垒逐渐消除,这些热门单抗的专利保护在2020年前后都相继到期,因此诸多企业不想错过这个大蛋糕,扎堆挤入了这个热潮。截至2016年3月,CDE受理的单抗药物申报,针对TNF-α、EGFR、VEGF、HER2这四个靶点已经有85个,占到单抗申报总数的75%以上(表2)。
一个热门单抗品种,数年后必然会有多个厂家产品上市,价格战必不可免。如同当初进口G-CSF制剂2000元/支到今天国产G-CSF 100+元/支一样,原来几万-几十万每年的费用必将下降一个数量级,这对社会和患者而言是一件幸事。因此,仿制单抗的未来必将跟现在诸多化学仿制药一样,逐渐由在资金,技术,品牌,政策等方面占据优势的中大型药企占据市场,小型star-up公司再去挤入开发仿制单抗的最佳时机已过。而创新药的开发,更多的是需要专业的技术团队、快速高效的决策机制、有效的团队激励体制,这些往往是成熟性药企的短板,因此小型生物企业去参与创新药物的开发反而具有更大的竞争优势。未来的中国生物医药研发模式会逐渐向西方靠拢:小公司靠技术生存,做前端的研发工作;大公司靠资金、规模、渠道优势,通过横向并购来获得更多的产品管线,更多聚焦于药品生产和销售领域。
3.2 “me-too创新药”的未来并非坦途
新药开发是一个全球竞争的格局,中国企业无论是技术、人才还是资金,跟国际制药巨头相比,都有着数量级的差距。如何在人才和资金都不占优势的情况下中国的生物医药研发能在全球竞争中立足,也是一件极具挑战的工作。在这种客观条件限制下,过去、现在及未来,国内一直存在一些学术上存在诸多争议的“me-too创新药”。
3.2.1 中国历史上曾经的“全球创新药”命运变化多端
回顾历史,中国在创新药物开发领域也曾做了不少探索,CFDA也曾批准过数个国际第一的创新药在中国上市。如全球唯一一个批准上市的Endostatin(商品名恩度),世界首个基因治疗药物“今又生”等等。这些在特殊历史条件下批准的创新药物,虽然批准上市时轰轰烈烈,最终因临床疗效等原因,逐渐回归寂寞。但是仍有一些产品,带着“创新药”的光环和独家品种的优势,继续在市场广泛流通。如“全球独家”的鼠源神经生长因子自2003年在中国批准上市,销售额持续增长,2016年销售额合计达到惊人的25.3亿元。但是这些制品和临床价值似乎不是那么让人信服,从科学性角度来说甚至有些令人匪夷所思。在逐渐强调药品临床价值的大趋势下,这些品种的未来走向如何,尚待观察。
3.2.2 “me-too创新药”过去的成功模式在未来不一定可以复制
真正的创新药物的研究不可能是一蹴而就的,受诸多因素的限制,中国的不少上市的“创新药”,除了临床疗效,其创新程度也颇受争议。如饶毅就曾发文质疑《我国今天的新药创新超过了1970年代吗?》。确实,近几年国内上市的有几个“创新药”就其创新程度和科学意义是值得商榷的,但是在原研药物上市慢、价格高的时候,他们在一定程度上满足了中国患者的药物可及性问题,因此其特定历史时期下的价值还是值得肯定的。因为中国特殊的药品审批政策所致,进口新药在中国的上市往往比国外要晚3-8年。有些进口药物即使上市了,每疗程数万至数十万的价格,让患者经常陷入望药兴叹的局面。而一些仿版“创新药”虽然在疗效上有时不能完全媲美原研药物,但是其更为亲民的价格,在一定程度上满足经济能力有限的患者的临床需求。这也是政府和社会对“康柏西普”、“埃克替尼”这类“me-too创新药”赞誉有加的一个重要原因。这些产品为患者提供了一定的临床疗效,市场也给予了足够的回报。贝达药业仅靠埃克替尼一个产品达到了300多亿的市值。受这些成功榜样的影响,大家投入开发这类fast follow-on产品,期待复制“康柏西普”,“埃克替尼”的辉煌。像目前最热门的PD1/PDL-1,已经报到CDE的已经有十多家(截至4月21日,国内注册申报的PD-1/PD-L1单抗药物共13个,其中包括9个PD-1单抗,4个PD-L1单抗[5]。这些fast follow-on的产品最后有几个能在临床试验中做出优效或非劣来呢?就算这些follow-on产品技术水平都不差,最终都能批准上市,那么十几个相同产品竞争的格局,有几个能成为赢家呢?别忘了“康柏西普”、“埃克替尼”是在国产独家的前提下才获得现在的市场份额和超额利润的。随着药审制度改革,进口药在国内上市速度会显著加速,国内新药的批准日益强调新的临床价值。因此,现在再去复制“康柏西普”、“埃克替尼”的模式,还是存在较大的不确定性的。
3.2.3 中国现有创新生物药研究水平还不高
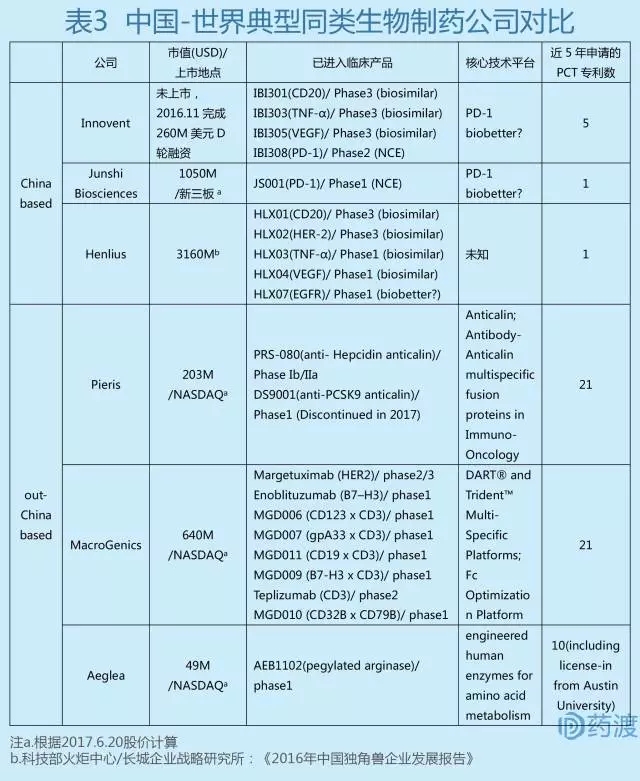
我们更要清醒地认识到,中国现有创新生物药研究水平跟世界相比还是有距离的。确实,近两年中国创新药研究领域热点颇多,巨额资本不断涌入,明星企业纷纷涌现,并已有数个产品许可给国际大药企,这是以前从未有过的现象。但是仔细评估国内生物药物研发的现状,我们还是认为国内生物医药的研究能力,特别是创新能力,与世界主流还是有一定差距的。表3对国内几个目前生物医药领域最具有代表性的明星企业与国外同类性质公司进行了简单比较分析。
从国内这些明星企业已经进入临床的产品管线来看,除生物仿制药外,集中于几个热门靶点的follow-on产品,尚未有first-in-class的产品。而且这些follow-on产品,从目前所披露的信息来看,跟第一代产品相比是否真正有临床优势,尚无法获知。另一方面,从最能衡量一个生物医药企业科技实力的PCT专利申请状态来看,国内企业的数量与质量均差强人意。从现有公开披露的信息来分析,除了投资额和硬件设施外,似乎很难判断国内这些公司核心技术优势在何处。而绝大部分国外生物医药研发小公司,创新程度非常高,而且都是具有自己独特的核心平台技术,然后基于这个核心技术去开发一些first-in-class或best-in-class产品。
3.3 寻找适合中国国情的创新发展道路
限于中国相对薄弱的基础研究,现阶段更多地聚焦于去开发fast follow-on及biobetter产品,应该是一条可行之路。对于一些确定很有前景的靶点,针对现有产品一些缺陷进行针对性改进,开发出真正具有更好临床意义的biobetter产品,会比做创新机制或者创新靶点成功的可能性大很多。生物药物的作用机制一般都非常明确,对于follow-on的生物药物,更有可能根据前一代产品的临床结果有针对性地进行改进并获得新的专利。药品是一类极其特殊的产品,对于创新靶点药物(first-in-class),即使已经批准上市,其结构与活性,临床疗效,副作用等因果关系在新药上市时往往并不非常明确,很多弊端或优势需要在大规模长期临床使用后才能暴露出来。因此针对同一靶点,往往出现第二代产品比第一代更成功的现象。例如,通过拮抗VEGF来治疗AMD,最早上市是Macugen,但是当疗效更好的Lucentis上市后,Macugen的市场迅速萎缩到退市边缘(见表4)。但是,罗氏在开发Lucentis的时候,为了提高所谓的组织渗透性,缩短全身半衰期,在Bevacizumab结构中截取了Fab片段。但是后面的临床Bevacizumab用于AMD同样安全有效,Bayer/Regeneron的Eylea保留了Fc,增大了分子量,凭借其更好的亲和力和更长的眼内PK,更少的给药频率却比Lucentis具有更好的疗效。因此Eylea虽然比Lucentis晚约5年上市(2012美国获批),但是到2015年,Eylea的销售已经超过Lucentis。PEG-G-CSF与G-CSF、PEG-IFN与IFN等等均是第二代产品的销售额远远大于第一代。国内目前一大批做PD-1、anti-PCSK9、TKIs抑制剂类药物采取的都是这个策略。问题的核心在于能不能在短期内真正获得一个等效甚至优效的follow-on产品。如果follow-on产品连临床等效都达不到,在现在的审批制度下能否批准上市?即使批准上市,能否能够占有足够的市场?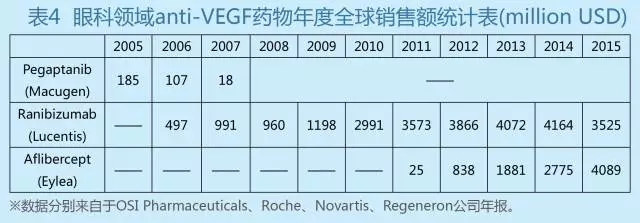
与简单的fast follow-on相比,采用新的技术去开发follow-on产品,从而真正获得biobetter药物,那么就属于创新药开发更高一个层次了。但是国内企业真正从事这方面研究的目前并不多。如未来有可能与单抗技术相竞争的scaffold技术,国外已经有Nanobody、Affibody、Darpins、Anticalin、Adnectins等十多个平台技术在研;基于全人工设计,用于代替PEG修饰技术的“recombinant Pegylation”,已经有XTEN、ELP、PASylation等不同的技术方案用于临床产品。再如现在炙手可热的bsAb领域,国外已经有BiTE,KiH、CrossMAB、DVD-Ig、Duobody等十多种技术平台。但是国内还很少有真正具有这类独特技术平台的公司。不过,我们也欣喜的看到至少有这个苗头。如岸迈生物(Epimab)的FIT-Ig双特异性抗体技术,道尔生物(Doer Biologics)的xLONG重组长效技术平台,武汉友芝友的YBODY双特异性抗体技术等。这些技术平台已经同步甚至优于国外同类技术,从开发成功率来看,基于创新技术平台的产品成功的概率并不一定高,但是一旦成功,可以基于这些技术平台开发出一系列真正的biobetter甚至best-in-class的创新药物。只有涌现出更多的这类真正拥有核心创新技术的小公司,中国才有可能开发出具有新的临床价值的创新药。
多靶点/组合药物创新,也是中国开发创新药物的一条可能道路。生物药物在诞生之初就带着“特异性好、副作用低”的光环,但是凡事都是两面的。人体是一个有机整体,大部分疾病本身并非单因素的作用。如果能够同时作用于疾病网络中多个靶点,对各靶点的作用产生协同效应,使总效应大于各单效应之和,往往能够达到最佳的治疗效果[6]。在化学药领域,多靶点药物已是常见。如多靶点口服抗癌药物索拉非尼(Sorafenib),能同时抑制VEGFR、PDGFR、FLT3、KIT受体酪氨酸激酶活性和Raf激酶。但是生物药物领域的多靶点/联合治疗药物却是近期才展露头角。在肿瘤免疫治疗领域,多靶点联合治疗目前正是炙手可热。如anti-PD-1与anti-CTLA-4联合治疗在黑色素瘤中体现的神奇疗效,是以往抗肿瘤药物从来没有达到过的。仅罗氏就开展了十多项Atezolizumab (anti-PD-1)联合治疗的临床试验。去年刚刚批准上市的Soliqua (甘精胰岛素100U/mL+利西拉来,iGlarLixi)和Xultophy (德谷胰岛素+利拉鲁肽,IDegLira)也是双靶点给药的一个成功组合。未来这样组合用药方案会越来越多。如Trastuzumab和Pertuzumab是针对Her2靶点不同表位的两个单抗,临床证实联合应用具有更好的疗效。因此Symphogen开发了系列混合抗体药物,如含两个anti-EGFR的Sym004,包含6个全长抗体的针对EGFR、HER2和HER3的Sym013等系列混合抗体药物。现有生物药物的组合方案几乎是无限的,特别是肿瘤免疫治疗领域的靶点组合更是层出不穷。但是这些组合是否有效,更是需要大量临床验证,即使是国际大公司,也只能选择少量组合去临床试验。因此针对中国病人的临床需求,去做一些多靶点组合药物,如果能够达到临床有效的效果,也是一种非常有价值的创新。
除了复方组合,更进一步的,针对一些单药疗效不好甚至单药无法成药的靶点,利用各种技术将多个活性结构域组合到一个单一分子结构中,也是开发创新药的一个方向。生物药物作用机制更为清楚,将多个活性结构域组合在一个分子上的难度也更低,因此更容易开发基于单一分子的多靶点药物。在全球领域这已经成为一个新的趋势,如罗氏公司目前处于3期临床研究前的生物药物中,已经有70%以上属于多结构域产品[7]。这种多结构域生物药已经越来越多的进入临床研究,仅仅基于bsAb机理的就已经有40多个产品处于临床研究中[8]。在糖尿病领域,也已经有十多个双/多重激动GLP-1、GIP、GCG多个受体的多效药物进入临床研究[9]。在国内也有少量比较有前瞻性的公司也早已介入了这个方向的研究。
3.4 引进创新,不一定是在中国开发创新药的捷径
国内小公司纷纷许可国外大企业临床中产品,成为近几年中国新药开发的一个新现象。如前所述,在中国创新药的研发其实还是与世界水平有比较大的距离的,但是因创新药物资产稀缺,创新药以及其研发公司价值在中国的估值是远远高于世界水平的。表1所列的对比可以明显看出这个估值差距。在国外,有1-2个first-in-class进入I期临床的公司,估值一般都不超过2亿美元;但是在中国,有1-2个热门fast follow-on 产品进入I期临床的公司,估值会超过10亿美元。如上海君实仅靠一个PD-1领先恒瑞1个月进入临床,市值达到了惊人的10.5亿美元。
大家看到了这个价值差异,开始了新的探索——直接收购国外的创新产品,到中国来继续后续开发甚至上市。应该说这确实也算是一条捷径,但是目前中国引进开发的很多案例又似乎有些与众不同。国际上新药并购的主流模式是从小公司流向大公司,一般都是小公司做前端,有进展了,产品或公司被大公司收购。但是在中国普遍是逆向的流动。大多数并购都是国内的新药研发小公司从国外大公司引进许可产品。创办人通过引入风险资本,通过技术许可迅速获得临床阶段的创新药物,迅速把一个start-up的小公司包装成一个拥有clinic-stage的创新药研发公司。再鼎医药(Zai lab)就是这种模式的一个典型。再鼎医药(Zai lab)自2014年成立后,分别从BMS许可了抗肝癌药“布立尼布”(Brivanib),从UCB许可了治疗自身免疫疾病的单抗,从赛诺菲许可了一个用于NSCLC的TKI和两种用于治疗慢性呼吸道疾病等多项创新药物,从韩美许可了用于肺癌的TKI (HM61713)。其他如华领从罗氏引进2型糖尿病新药Sinogliatin(葡萄糖激酶激活剂,GKA),杭州歌礼生物从罗氏引进的丙肝药物Danoprevir,派格从辉瑞引进的葡萄糖激酶激活剂(GKA),2016年12月天境生物引进辉凌制药(Ferring Pharma)的Olamkicept(白介素IL-6通路抑制剂)(在亚洲地区针对自身免疫类适应症的开发和商业化的独家授权),都是这样的例子。
在中国极度缺乏创新药资产和概念而资本又严重过剩的环境下,这种模式确实颇受资本市场欢迎,也有可能是创新药研发的一条捷径。但是,靠购买产品而非技术,不建立自己的研发体系,全部依靠外包这种开发模式,永远只是一种商业模式,很难真正建立起核心技术平台,也很难有自己可持续发展的pipeline。其次,更大疑问还是产品本身。如果不是因为对这些产品失去了信心,很难想象国际制药巨头为什么会把产品许可给中国这些技术和渠道都是空白的小公司。那么,被大药企放弃的创新药真的有可能再获得批准么?像Brivanib、GKA这些已经在国外临床中被放弃的产品,其最终的临床数据最终能否说服CFDA?CFDA敢不敢冒这个风险去批准像Brivanib这种已经明确疗效不如索拉非尼的产品?中国企业去引进这些产品,并不一定真的是认为在国内去做临床能够获得比原来更好的结果,更可能出于以下一些考虑:①国外上市原研产品价格昂贵,因此开发一个疗效比原研略差但是价格低很多的新药,在中国还是有很大市场,埃克替尼就是一个成功的例子;②进口药物在中国审批周期长,进口原研药物在中国上市时间远远落后于国外,通过引进产品,利用国内大量病人资源,快速完成临床研究,甚至有可能实现比原研更快上市而先行一步抢占市场。但是,2017年3月17日,CFDA发布《关于调整进口药品注册管理有关事项的决定(征求意见稿)》的出台,为这类引进产品增加了一些变数。若该规定最后实施,将大幅缩短进口新药在中国上市速度。国外药品从I期就可以开始在中国进行临床试验,完成了包括中国在内的国际多中心临床试验后,可以直接提出上市注册申请。国外原研产品从原来的三报三批,上市的进程缩短2-5年。因此这些引进中国的follow-on制品,有可能完全丧失速度优势,如果产品本身在临床疗效体现不了优势,那么未来在中国获批上市的风险陡增。
4、外围环境的因素对生物创新药物开发的影响
4.1 CFDA的改革是否能跟上中国药物开发模式的转换节奏
在以往仿制药开发为主的模式下,在中国一直是批准进临床试验难,批准上市生产容易。这种模式是无法满足创新药,特别是创新生物药的开发需求的。生物药物不同于化药和中药,由于存在种属差异,很多生物药物的有效性和安全性根本无法在临床前动物试验中体现,特别是像CAR-T和T-细胞重定向双特异抗体[10]类产品,目前很难有合适的动物模型,只有在人体试验中才能去真正开展有效性和安全性研究,其技术挑战和潜在风险不言而喻。因此国内越来越多的创新生物药临床申请对CFDA审批的科学性和勇气提出了更高的要求。
从这几年国家药品管理相关法规改革和实践来看,国内药品的审批和监管措施逐渐变得科学合理。最近奥希替尼从受理(2016年9月)到批准上市(2017年3月)仅仅花了6个月,这个惊人的速度真正体现了CFDA朝着科学监管的方向发展的决心。近一年密集发布的各种新的审批政策更是体现了这种转变。在2017年5月11日一天发布了3个征求意见稿[11-13],提出了缩短创新药的审批时限,取消了临床机构的认证要求等诸多重大政策改革措施。
但是,如果真的中国新药研发的重心开始朝着真正创新药物方向发展,对于CFDA也存在不小的挑战。专业审批技术人员紧缺就是CFDA首先要面临的一个现实问题。虽然自2016年起,CDE已经在大幅招兵买马,但是这方面的专业人才在国内本来就奇缺,跟众多财大气粗的企业相比,在CDE做审评工作,任务繁重,责任重大但待遇有限,真正要吸引大量专业经验丰富的人士进入并留在CDE,并非易事。其次,原有的审批体制惯性巨大,CFDA是否真正能在短期内摆脱原有的约束,真正走向科学审评,科学监管的道路,尚有待时间的考验。现行的新药审批责任终身制,使得每个负有审批职责的官员都变得非常谨慎,难免会有“不求有功,但求无过”的思维[14]。这样的机制下,更容易批准的是一些安全无效的创新药物。虽然一再要求创新药“以满足了尚未满足的临床需求”为标准,仍然在2016年12月份批准了贝那鲁肽注射液(重组GLP-1,每天注射3次)这种颇具争议的创新药上市,更显得CFDA的改革道路任重道远。
4.2 临床资源和技术能力会是未来国内创新药物开发的一个限速环节
真正的创新药物开发,不是仅仅依靠实验室的研究,更需要大量的临床试验来决定其真正的有效性和安全性。中国创新药物开发的热潮,也必然导致临床资源的紧缺,这可能是未来影响中国创新药物的研发效率的重要瓶颈。中国人口众多,临床病人资源并不缺乏,但是在临床研究基地的容量和能力两方面都存在严重的不足和挑战。一方面是临床基地数量的不足,由于监管机构对于临床研究的重视程度不足,比如公立医院监管的主体,并没有把临床研究当做是评估三甲医院的关键指标,在国内能够开展临床研究的床位数与人口总数是严重不符的。其次是开展临床研究能力与国际水平有较大差距,特别是创新型临床研究能力更为缺乏。中国以往的新药开发模式决定了绝大部分医院和临床医生缺乏创新药物早期临床的经验和能力。除了技术能力的缺乏,中国紧张的医患关系,也为临床医生开展临床试验的积极性带来了额外的忧虑。毕竟完全创新机制的新药临床,特别是生物创新药的临床,其难度和不可预见性不是以前那些“新药”临床试验相比的。如果进口药品在中国临床注册程序进行改革,国外开发的新药可以选择中国进行I期临床研究,那么本来就奇缺的创新药临床资源必然更为稀缺。国内创新药开发企业在I期临床阶段就将必须与国际巨头争夺临床资源,对资金和技术都不占优势的国内新药开发企业来说又是一个挑战。
5、结语
总之,现有的中国临床用药现状还是与世界主流相脱节的,一些无效安全的药品还占据了很大一部分临床市场,无谓消耗了大量社会资源,生物药物在中国目前的份额还非常小。但是我们可以看到现状已经逐渐在改变,国家的监管政策日趋科学,一些高风险、无临床收益的产品在逐步被驱出市场,以临床价值为核心将逐渐成为中国药品市场的主流。因此临床价值巨大的生物药物必将逐渐在未来中国的药品市场占据重要分量。但是现有的科研基础,政策环境,外部资源等等诸多因素的限制,中国的生物药物产业何时赶上世界前沿,只需要时间来验证。
参考文献
[1] Preetika Rana. China Emerges as Powerhouse for Biotech Drugs[N]. Wall StreetJournal,2017-4-10.
[2] 卢敏丽. 2016年中国医药市场格局大盘点. 第76届全国药品交易会. 2016年.
[3] Zhang M,Zhang J,Yan M,et al.Recombinant anti-vascular endothelial growth factor fusion protein efficiently suppresses choridal neovasularization in monkeys[J]. Mol Vis,2008,14(1): 37-49.
[4] Nguyen T T,Guymer R. Conbercept (KH-902) for the treatment of neovascularage-related macular degeneration[J]. Expert review of clinical pharmacology,2015,8(5): 541-548.
[5] 医药魔方. 国内药企PD-1申报情况、开发进度、临床试验信息汇总[EB/OL]. http://www.pharmcube.com/news/article/2372,2017-4-23.
[6] Zimmermann G R,Lehar J,Keith C T. Multi-target therapeutics: when the whole is greater than the sum of the parts[J]. Drug discovery today,2007,12(1):34-42.
[7] Stefan Weigand. Fusion Proteins: Introduction to the Field and Case Studiesfrom Roche’s Research & Early Development Pipeline. PEGS: The Essential Protein Engineering Summit,Boston,America,April 25-29,2016.
[8] Trivedi A,Stienen S,Zhu M,etal. Clinical Pharmacology and Translational Aspects of Bispecific Antibodies[J]. Clinical and Translational Science,2017.
[9] Tschöp M H,Finan B,Clemmensen C,et al. Unimolecular polypharmacy for treatment of diabetes and obesity[J]. Cell metabolism,2016,24(1): 51-62.
[10] Zhukovsky E A,Morse R J,Maus M V. Bispecific antibodies and CARs: generalized immunotherapeutics harnessing Tcell redirection[J]. Current opinion in immunology,2016,40: 24-35.
[11] 国家食品药品监督管理总局. 总局关于征求《关于鼓励药品医疗器械创新加快新药医疗器械上市审评审批的相关政策》(征求意见稿)意见的公告(2017年第52号)[EB/OL]. http://www.sda.gov.cn/WS01/CL0087/172567.html,2017-5-11.
[12] 国家食品药品监督管理总局.总局关于征求《关于鼓励药品医疗器械创新改革临床试验管理的相关政策》(征求意见稿)意见的公告(2017年第53号)[EB/OL].http://www.sda.gov.cn/WS01/CL0087/172568.html,2017-5-11.
[13] 国家食品药品监督管理总局.总局关于征求《关于鼓励药品医疗器械创新实施药品医疗器械全生命周期管理的相关政策》(征求意见稿)意见的公告(2017年第54号)[EB/OL].http://www.sda.gov.cn/WS01/CL0778/172569.html,2017-5-11.
[14] 樊纲. 破除新药审评人员责任终身制,是发展我国生命健康产业的当务之急[EB/OL]. http://www.50forum.org.cn/home/article/detail/id/6756.html,2016-12-12.
作者介绍
黄岩山,浙江道尔生物科技有限公司创始人/CEO,致力于建立创新的 “biobetter” 及 “best-in-class” 类长效多结构域技术平台。曾任杭州九源基因工程有限公司首席科学家兼研究所所长,参与创建了江苏泰康生物医药有限公司,任公司高级副总裁。建立了长效蛋白药物和抗体药物两个技术平台,完成了3个创新生物药物和1个生物仿制单抗药的开发,目前均处于不同临床阶段。